Planning with RDKit#
This tutorial shows how to use SynPlanner’s retrosynthetic planning with RDKit Mol objects for input and output. If you’re comfortable with RDKit and want to use SynPlanner without learning chython, this is the place to start.
The workflow:
Load reaction rules and policy network (SynPlanner-native, no changes needed)
Create your target molecule with RDKit
Convert it for SynPlanner with one function call
Run the tree search
Get results back as RDKit Mol objects
For a deeper introduction to chython and how it relates to RDKit, see Tutorial 01: Coming from RDKit.
1. Download preset data#
SynPlanner provides pre-trained models and building blocks. We download them once.
[ ]:
from pathlib import Path
from synplan.utils.loading import download_preset
paths = download_preset("synplanner-article", save_to="synplan_data")
ranking_policy_network = paths["ranking_policy"]
reaction_rules_path = paths["reaction_rules"]
building_blocks_path = paths["building_blocks"]
2. Load reaction rules, policy, and building blocks#
These are loaded with SynPlanner’s standard functions. Nothing RDKit-specific here.
[ ]:
from synplan.utils.loading import (
load_building_blocks,
load_reaction_rules,
load_policy_function,
load_evaluation_function,
)
building_blocks = load_building_blocks(building_blocks_path, standardize=True, silent=False)
reaction_rules = load_reaction_rules(reaction_rules_path)
policy_function = load_policy_function(weights_path=ranking_policy_network)
print(f"Loaded {len(building_blocks):,} building blocks")
print(f"Loaded {len(reaction_rules)} reaction rules")
3. Create target molecule with RDKit#
Use your familiar RDKit workflow to create and inspect the target molecule.
[ ]:
from rdkit import Chem
from rdkit.Chem import Draw, Descriptors
# Capivasertib — anti-cancer medication approved by FDA in 2023
target_smiles = "NC1(C(=O)N[C@@H](CCO)c2ccc(Cl)cc2)CCN(c2nc[nH]c3nccc2-3)CC1"
rdkit_target = Chem.MolFromSmiles(target_smiles)
print(f"Molecular weight: {Descriptors.ExactMolWt(rdkit_target):.1f}")
print(f"Heavy atoms: {rdkit_target.GetNumHeavyAtoms()}")
Draw.MolToImage(rdkit_target, size=(400, 300))
4. Convert target for SynPlanner#
target_from_rdkit converts an RDKit Mol to the internal representation and applies standardization.
[ ]:
from synplan.chem.rdkit_compat import target_from_rdkit
target = target_from_rdkit(rdkit_target)
# Side-by-side: RDKit type vs chython type
print(f"RDKit type: {type(rdkit_target).__name__}")
print(f"Chython type: {type(target).__name__}")
print(f"Canonical SMILES: {str(target)}")
# chython depiction (works in Jupyter)
target
(Optional) Custom building blocks from RDKit#
If you have a custom set of building blocks as RDKit Mol objects — for example, molecules standardized via your RDKit pipeline — you can convert them directly:
[ ]:
from synplan.chem.rdkit_compat import building_blocks_from_rdkit
# Example: a small custom set of building blocks
custom_bb_smiles = ["CCO", "CC(=O)O", "c1ccccc1", "OCC"]
custom_rdkit_bbs = [Chem.MolFromSmiles(s) for s in custom_bb_smiles]
custom_bb_set = building_blocks_from_rdkit(custom_rdkit_bbs)
print(f"Input: {len(custom_bb_smiles)} SMILES -> {len(custom_bb_set)} unique building blocks")
for smi in sorted(custom_bb_set):
print(f" {smi}")
# For this tutorial, we use the downloaded building blocks
# custom_bb_set would be passed to Tree(building_blocks=custom_bb_set)
5. Configure and run tree search#
Standard SynPlanner configuration. The converted target molecule is passed directly to the Tree.
[ ]:
from synplan.utils.config import TreeConfig, RolloutEvaluationConfig
tree_config = TreeConfig(
search_strategy="expansion_first",
max_iterations=300,
max_time=120,
max_depth=9,
min_mol_size=1,
init_node_value=0.5,
ucb_type="uct",
c_ucb=0.1,
)
eval_config = RolloutEvaluationConfig(
policy_network=policy_function,
reaction_rules=reaction_rules,
building_blocks=building_blocks,
min_mol_size=tree_config.min_mol_size,
max_depth=tree_config.max_depth,
)
evaluation_function = load_evaluation_function(eval_config)
[ ]:
from synplan.mcts.tree import Tree
tree = Tree(
target=target,
config=tree_config,
reaction_rules=reaction_rules,
building_blocks=building_blocks,
expansion_function=policy_function,
evaluation_function=evaluation_function,
)
for solved, node_ids in tree:
pass
tree
6. Extract routes as RDKit objects#
Two export functions are available:
``route_to_rdkit(tree, node_id)`` — flat list of retrosynthetic steps for a single route
``extract_routes_rdkit(tree)`` — nested tree structure for all winning routes
Both return RDKit Mol objects with atom mapping preserved by default.
6.1 Flat step list#
[ ]:
from synplan.chem.rdkit_compat import route_to_rdkit
# Pick the best route
best_node = tree.winning_nodes[0]
steps = route_to_rdkit(tree, best_node)
print(f"Route has {len(steps)} steps:\n")
for i, step in enumerate(steps):
target_smi = Chem.MolToSmiles(step['target'])
print(f"Step {i + 1} (rule {step['rule_id']}, depth {step['depth']}):")
print(f" Target: {target_smi}")
for mol, stock in zip(step['precursors'], step['in_stock']):
status = 'in stock' if stock else 'needs synthesis'
print(f" -> {Chem.MolToSmiles(mol)} ({status})")
print()
6.2 Visualize steps with RDKit#
Since the results are RDKit Mol objects, you can use any RDKit visualization.
[ ]:
from rdkit.Chem import AllChem, Draw
# Collect all unique molecules from the route
all_mols = []
all_legends = []
for i, step in enumerate(steps):
all_mols.append(step['target'])
all_legends.append(f"Step {i+1} target")
for j, (mol, stock) in enumerate(zip(step['precursors'], step['in_stock'])):
all_mols.append(mol)
status = 'BB' if stock else ''
all_legends.append(f"Step {i+1} precursor {j+1} {status}")
# Remove atom mapping and recompute 2D coords for clean depiction
clean_mols = []
for mol in all_mols:
mol_copy = Chem.RWMol(mol)
for atom in mol_copy.GetAtoms():
atom.SetAtomMapNum(0)
mol_copy = mol_copy.GetMol()
AllChem.Compute2DCoords(mol_copy)
clean_mols.append(mol_copy)
Draw.MolsToGridImage(
clean_mols,
legends=all_legends,
molsPerRow=4,
subImgSize=(300, 250),
)
6.3 Nested route tree#
extract_routes_rdkit returns the full route tree structure with RDKit Mol objects at every node.
[14]:
from synplan.chem.rdkit_compat import extract_routes_rdkit
routes = extract_routes_rdkit(tree)
print(f"Found {len(routes)} routes\n")
# Print the first route's tree structure
def print_route_tree(node, indent=0):
prefix = " " * indent
if node["type"] == "mol":
status = "[BB]" if node["in_stock"] else ""
smi = Chem.MolToSmiles(node["mol"])
print(f"{prefix}{smi} {status}")
elif node["type"] == "reaction":
print(f"{prefix}=>")
for child in node.get("children", []):
print_route_tree(child, indent + 1)
print_route_tree(routes[0])
Found 147 routes
[NH2:1][C:2]1([C:3](=[O:4])[NH:5][CH:6]([CH2:7][CH2:8][OH:9])[c:10]2[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]2)[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1
=>
[NH:1]([C:2]1([C:3](=[O:4])[NH:5][CH:6]([CH2:7][CH2:8][OH:9])[c:10]2[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]2)[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37]
=>
[NH:1]([C:2]1([C:3](=[O:4])[NH:5][CH:6]([CH2:7][C:8](=[O:9])[O:38][CH3:39])[c:10]2[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]2)[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37]
=>
[NH:1]([C:2]1([C:3](=[O:4])[OH:40])[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37]
=>
[NH:1]([C:2]1([C:3](=[O:4])[OH:40])[CH2:17][CH2:18][NH:19][CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37]
=>
[NH:1]([C:2]1([C:3](=[O:4])[OH:40])[CH2:17][CH2:18][N:19]([C:41](=[O:42])[O:43][CH2:44][c:45]2[cH:46][cH:47][cH:48][cH:49][cH:50]2)[CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37] [BB]
[c:20]1([Cl:41])[n:21][cH:22][n:23][c:24]2[nH:25][cH:26][cH:27][c:28]12 [BB]
[NH2:5][CH:6]([CH2:7][C:8](=[O:9])[O:38][CH3:39])[c:10]1[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]1 [BB]
7. Build RDKit ChemicalReaction from steps#
Since atom mapping is preserved, you can construct proper RDKit ChemicalReaction objects from the step molecules.
[15]:
from rdkit.Chem import AllChem
step = steps[0] # first retrosynthetic step
# Build reaction SMILES: precursors >> target (forward synthesis direction)
precursor_smiles = ".".join(Chem.MolToSmiles(m) for m in step["precursors"])
target_smiles_rxn = Chem.MolToSmiles(step["target"])
rxn_smiles = f"{precursor_smiles}>>{target_smiles_rxn}"
print(f"Reaction SMILES: {rxn_smiles}\n")
rxn = AllChem.ReactionFromSmarts(rxn_smiles, useSmiles=True)
print(f"Reactants: {rxn.GetNumReactantTemplates()}")
print(f"Products: {rxn.GetNumProductTemplates()}")
Draw.ReactionToImage(rxn, subImgSize=(300, 200))
Reaction SMILES: [NH:1]([C:2]1([C:3](=[O:4])[NH:5][CH:6]([CH2:7][CH2:8][OH:9])[c:10]2[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]2)[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1)[C:31]([O:32][C:33]([CH3:34])([CH3:35])[CH3:36])=[O:37]>>[NH2:1][C:2]1([C:3](=[O:4])[NH:5][CH:6]([CH2:7][CH2:8][OH:9])[c:10]2[cH:11][cH:12][c:13]([Cl:14])[cH:15][cH:16]2)[CH2:17][CH2:18][N:19]([c:20]2[n:21][cH:22][n:23][c:24]3[nH:25][cH:26][cH:27][c:28]23)[CH2:29][CH2:30]1
Reactants: 1
Products: 1
[15]:
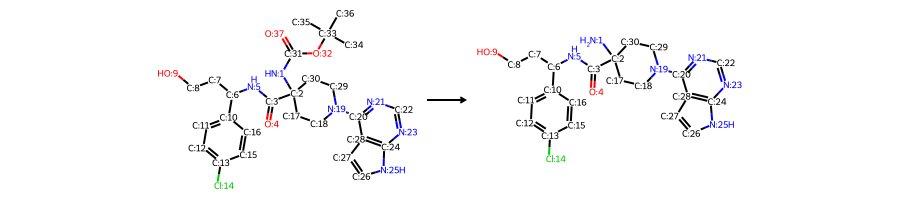
8. Analyze precursors with RDKit descriptors#
Since all molecules are RDKit objects, you can compute any RDKit descriptor or fingerprint.
[16]:
from rdkit.Chem import Descriptors
print(f"{'SMILES':<50} {'MW':>8} {'LogP':>8} {'TPSA':>8} {'Stock':>6}")
print("-" * 86)
for step in steps:
for mol, stock in zip(step['precursors'], step['in_stock']):
# Remove atom mapping for clean SMILES
mol_clean = Chem.RWMol(mol)
for atom in mol_clean.GetAtoms():
atom.SetAtomMapNum(0)
mol_clean = mol_clean.GetMol()
smi = Chem.MolToSmiles(mol_clean)
mw = Descriptors.ExactMolWt(mol_clean)
logp = Descriptors.MolLogP(mol_clean)
tpsa = Descriptors.TPSA(mol_clean)
print(f"{smi:<50} {mw:>8.1f} {logp:>8.2f} {tpsa:>8.1f} {'yes' if stock else 'no':>6}")
SMILES MW LogP TPSA Stock
--------------------------------------------------------------------------------------
CC(C)(C)OC(=O)NC1(C(=O)NC(CCO)c2ccc(Cl)cc2)CCN(c2ncnc3[nH]ccc23)CC1 528.2 3.71 132.5 no
COC(=O)CC(NC(=O)C1(NC(=O)OC(C)(C)C)CCN(c2ncnc3[nH]ccc23)CC1)c1ccc(Cl)cc1 556.2 3.90 138.5 no
CC(C)(C)OC(=O)NC1(C(=O)O)CCN(c2ncnc3[nH]ccc23)CC1 361.2 1.91 120.4 no
COC(=O)CC(N)c1ccc(Cl)cc1 213.1 1.90 52.3 yes
CC(C)(C)OC(=O)NC1(C(=O)O)CCNCC1 244.1 0.72 87.7 no
Clc1ncnc2[nH]ccc12 153.0 1.61 41.6 yes
CC(C)(C)OC(=O)NC1(C(=O)O)CCN(C(=O)OCc2ccccc2)CC1 378.2 2.77 105.2 yes
9. Comparison: chython vs RDKit depiction#
For reference, here’s how the same route looks using SynPlanner’s built-in chython visualization.
[17]:
from IPython.display import SVG, display
from synplan.utils.visualisation import get_route_svg
print("Chython route visualization:")
display(SVG(get_route_svg(tree, best_node)))
Chython route visualization:

Summary#
The key functions from synplan.chem.rdkit_compat:
Function |
Purpose |
|---|---|
|
Convert RDKit Mol to search target |
|
Convert RDKit Mol list to building block set |
|
Export one route as flat step list with RDKit Mols |
|
Export all routes as nested tree with RDKit Mols |
All output functions preserve atom mapping by default (keep_mapping=True), which is required to build proper RDKit ChemicalReaction objects.
For more about chython and its relationship to RDKit, see Tutorial 01: Coming from RDKit.